AlphaFold neural network produced a ‘totally transformative’ database of more than 350,000 structures from Homo sapiens and 20 model organisms.
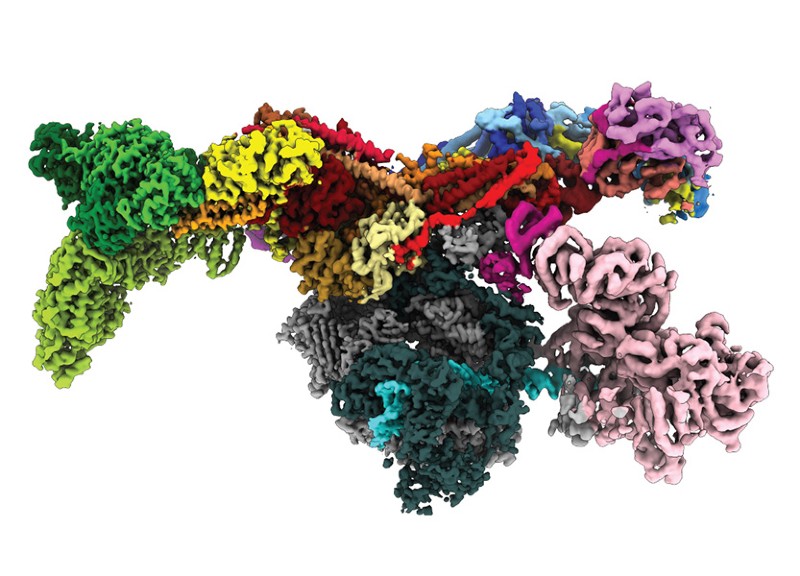
The human mediator complex has long been one of the most challenging multi-protein systems for structural biologists to understand.
19 July 2021 – The human genome holds the instructions for more than 20,000 proteins. But only about one-third of those have had their 3D structures determined experimentally. And in many cases, those structures are only partially known.
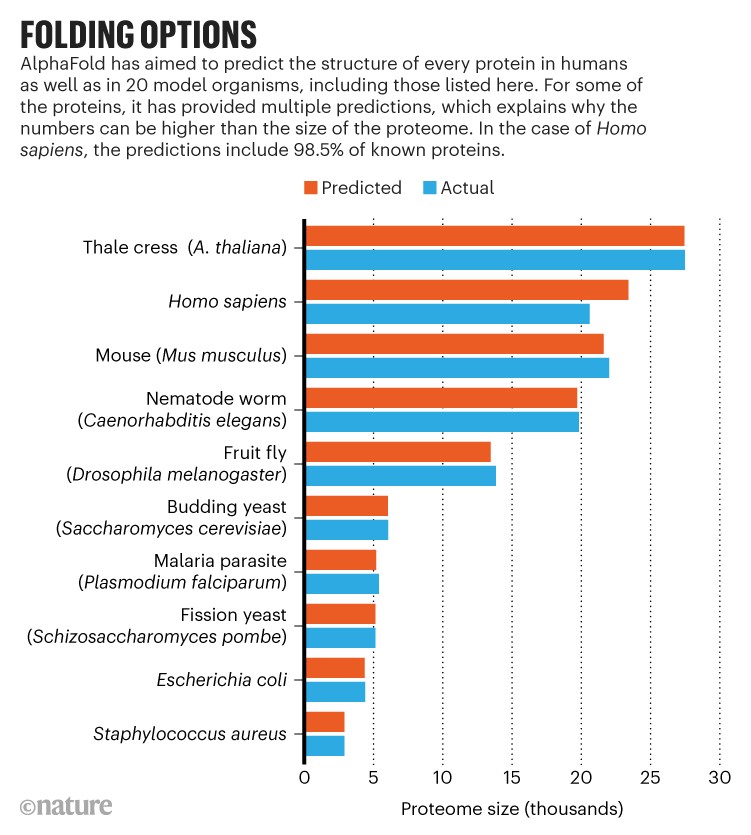
Now, a transformative artificial intelligence (AI) tool called AlphaFold, which has been developed by Google’s sister company DeepMind in London, has predicted the structure of nearly the entire human proteome (the full complement of proteins expressed by an organism). In addition, the tool has predicted almost complete proteomes for various other organisms, ranging from mice and maize (corn) to the malaria parasite.
The more than 350,000 protein structures, which are available through a public database, vary in their accuracy. But researchers say the resource — which is set to grow to 130 million structures by the end of the year — has the potential to revolutionize the life sciences.
“It’s totally transformative from my perspective. Having the shapes of all these proteins really gives you insight into their mechanisms,” says Christine Orengo, a computational biologist at University College London (UCL).
“This is the biggest contribution an AI system has made so far to advancing scientific knowledge. I don’t think it’s a stretch to say that,” says Demis Hassabis, co-founder and chief executive of DeepMind.
But researchers emphasize that the data dump is a beginning, not an end. They will want to validate the predictions and, more importantly, apply them to experiments that were hitherto impossible. “It’s an amazing first step, that we have all this data on that scale,” says David Jones, a UCL computational biologist who advised DeepMind on an earlier iteration of AlphaFold.
Prizewinning predictions
DeepMind stunned the life-sciences community last year, when an updated version of AlphaFold swept a biennial protein-prediction exercise called CASP (Critical Assessment of Protein Structure Prediction). In this long-running competition, which has traditionally been the domain of academics, researchers predict the structures of proteins whose structures have been experimentally solved, but not yet made public.
Some of AlphaFold’s predictions were on par with very good experimental models, and some scientists said the network’s influence would be epochal. Last week, DeepMind released the source code behind the latest version of AlphaFold, and a detailed description of how it was developed (academic teams have already begun using these resources to make useful predictions). In the process of preparing AlphaFold’s code for public release, DeepMind refined it to make the code run more efficiently. Some of the CASP predictions took days, but the updated version of AlphaFold could now compute them in minutes to hours.
With this added efficiency, the DeepMind team set out to predict the structures of nearly every known protein encoded by the human genome, as well as those of 20 model organisms. The structures are available in a database maintained by EMBL-EBI (the European Molecular Biology Laboratory European Bioinformatics Institute) in Hinxton, UK.
In addition to the predicted structures, which cover 98.5% of known human proteins and a similar percentage for other organisms, AlphaFold generated a measurement of the confidence of its predictions. “We want to give experimentalists and biologists a really clear signal of which parts of the predictions they should rely on,” says Kathryn Tunyasuvunakool, a science engineer at DeepMind and first author of a Nature paper describing the human proteome predictions. For the human proteome, 58% of its predictions for the locations of individual amino acids were good enough to be confident in the shape of the protein’s folds, Tunyasuvunakool says. A subset of those predictions — 36% of the total — are potentially precise enough to detail atomic features useful for drug design, such as the active site of an enzyme.
Even the less-accurate predictions might offer insights. Biologists think that a large proportion of human proteins and those of other eukaryotes — organisms with cells that have nuclei — contain regions that are are inherently disordered and take on a defined structure only in concert with other molecules. “Many proteins are just wiggly in solution, they don’t have a fixed structure,” says AlphaFold lead researcher John Jumper. Some of the regions that AlphaFold predicted with low confidence match up with those that biologists suspect are disordered, says Pushmeet Kohli, head of AI for science at DeepMind.
Determining how individual proteins interact with other cellular players is one of the greatest challenges to the AlphaFold predictions, say researchers. For the CASP competition, most of its predictions were of independently folding units of a protein, called domains. But the human proteome, and those of other organisms, contains proteins with multiple domains that fold semi-independently. Human cells also contain molecules made of multiple chains of interacting proteins, such as receptors on cell membranes.
Deluge of data
The approximately 365,000 structure predictions deposited this week should swell to 130 million — nearly half of all known proteins — by the year’s end, says Sameer Velankar, a structural bioinformatician at EMBL-EBI. The database will be updated as new proteins are identified and predictions improved. “This is not a resource you expect to have access to,” says Tunyasuvunakool, and she is eager to see what scientists come up with.
Researchers are already using AlphaFold and related tools to help make sense out of experimental data generated using X-ray crystallography and cryo-electron microscopy. Marcelo Sousa, a biochemist at the University of Colorado Boulder, used AlphaFold to make models from X-ray data of proteins that bacteria use to evade an antibiotic called colistin. The parts of the experimental model that differed from the AlphaFold prediction were typically regions that the software had assigned with low confidence, Sousa notes, a sign that AlphaFold is accurately predicting its limits.
Still, biologists will want to continue benchmarking these predictions to experimental data to get a better handle on their reliability, says Venki Ramakrishnan, a structural biologist at the MRC Laboratory of Molecular Biology in Cambridge, UK. “We need to be able to trust these data,” adds Orengo.
Jones is impressed with what the network has achieved. But he says that many of the models predicted by AlphaFold could have been generated with earlier software developed by academics. “For most proteins, those results are probably good enough for quite a lot of the things you want to do.” Scientists dead-set on obtaining the structure of any particular protein could probably succeed using experimental approaches.
But the availability of so many protein structures is likely to mark a “paradigm shift” in biology, says Mohammed AlQuraishi, a computational biologist at Columbia University in New York City who works on protein-structure prediction. His field has spent so much time and energy on predicting accurate protein structures on this scale that it hasn’t yet worked out what do with such resources. “Everything we do today that relies on a protein sequence, we can now do with protein structure.”
Orengo hopes that the database will help her to better understand the structural constraints of proteins. She has mapped a database of known proteins into about 5,000 ‘structural families’, but about half of the proteins in the database are excluded because there is nothing else like them for which a structure has been determined. AlphaFold’s predictions could help uncover new shapes, she says. “We’ll really see what folding space looks like.”
Jones expects AlphaFold will lead to a lot of soul-searching among biologists about what to do with so many structures — and the ease of creating many more. “There will be conferences. Now we’ve got 130 million models, how does this change our view of biology? It may be it doesn’t change it,” he says. “I suspect it will.”